Unveiling the Enigma of Vogt-Koyanagi-Harada Syndrome
Introduction
Vogt-Koyanagi-Harada (VKH) syndrome, though rare, manifests as a complex autoimmune disorder affecting multiple systems, notably the eyes, skin, auditory apparatus, and central nervous system. Its intricate interplay of symptoms and underlying immune dysfunction poses diagnostic and therapeutic challenges. In this comprehensive article, we embark on a detailed journey through the various facets of VKH syndrome, unraveling its clinical presentation, etiology, diagnostic modalities, and therapeutic interventions.
Understanding Vogt-Koyanagi-Harada Syndrome
VKH syndrome stands as a testament to the body’s immune system turning against its own tissues, particularly those containing melanocytes. These cells, responsible for pigment production, become the focal point of autoimmune attack, resulting in widespread inflammation and tissue damage across affected organs. Although the exact trigger remains elusive, genetic predisposition, coupled with environmental factors like viral infections, are believed to ignite the autoimmune cascade.
Symptoms of VKH Syndrome
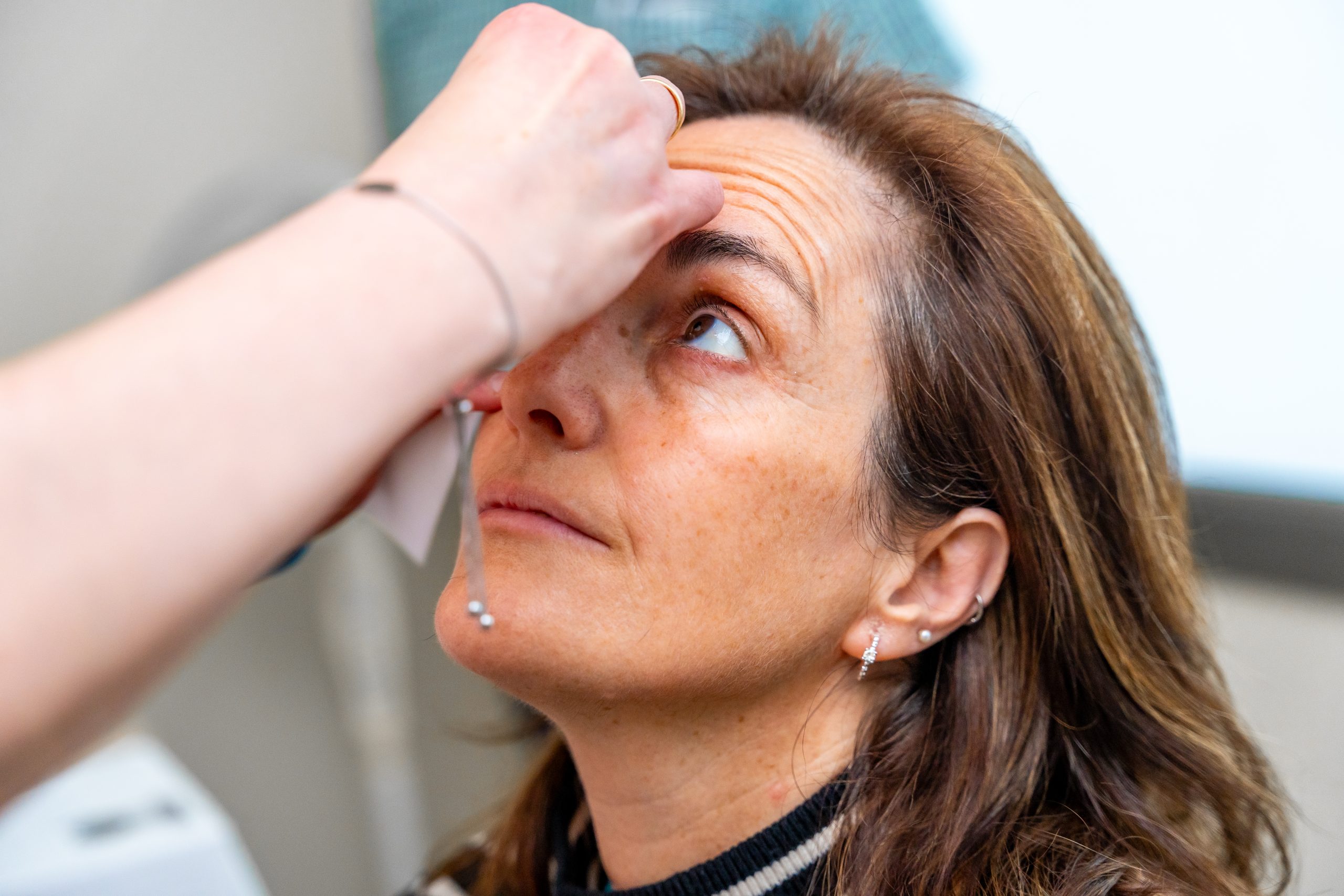
The clinical course of VKH syndrome unfolds in distinct stages, each marked by a spectrum of symptoms. The prodromal phase may commence with nonspecific manifestations akin to flu-like symptoms, including fever, headache, and general malaise. As the disease progresses, ocular involvement becomes prominent, with patients experiencing a myriad of visual disturbances:
- Blurred vision, stemming from inflammatory changes in the eye.
- Photophobia, rendering the eyes hypersensitive to light stimuli.
- Ocular pain, indicative of underlying inflammation within the eye structures.
- Redness and inflammation of the eyes (uveitis), manifesting as eye redness and discomfort.
- Visual distortions or loss, arising from retinal involvement or optic nerve inflammation.
- Presence of floaters, characterized by dark spots or lines in the visual field, signaling vitreous inflammation.
Beyond ocular manifestations, VKH syndrome casts its shadow over other organ systems, leading to skin depigmentation, hair loss, and auditory disturbances, such as tinnitus or hearing loss, underscoring its systemic nature.
Causes of VKH Syndrome
Unraveling the etiology of VKH syndrome involves navigating through a labyrinth of genetic predispositions and environmental triggers. Genetic susceptibility, underscored by specific human leukocyte antigen (HLA) haplotypes, predisposes individuals to immune dysregulation. Concurrently, environmental factors, ranging from viral infections like Epstein-Barr virus to traumatic events, serve as precipitating factors, inciting the autoimmune response against melanocytes.
Diagnosis of VKH Syndrome
Accurate diagnosis of VKH syndrome necessitates a meticulous approach, involving a confluence of clinical assessment, ophthalmic evaluation, and ancillary investigations. Key components of the diagnostic armamentarium include:
- Thorough medical history, encompassing symptom onset, systemic manifestations, and past medical conditions.
- Comprehensive ophthalmic examination, encompassing visual acuity assessment, intraocular pressure measurement, and slit-lamp biomicroscopy to detect ocular inflammation.
- Ancillary imaging modalities, such as optical coherence tomography (OCT), fluorescein angiography (FA), and indocyanine green angiography (ICGA), offering insights into retinal and choroidal changes.
- Laboratory investigations, including autoimmune serology panels and genetic testing, aid in corroborating the diagnosis and ruling out mimicking conditions.
Treatment Strategies for VKH Syndrome
Navigating the therapeutic landscape of VKH syndrome entails a multidisciplinary approach aimed at halting the autoimmune assault, mitigating inflammation, and averting disease recurrence. Treatment modalities encompass a triad of pharmacological agents and surgical interventions:
- Systemic corticosteroids serve as the cornerstone of therapy, exerting potent anti-inflammatory effects to quell ocular and systemic inflammation.
- Immunosuppressive agents, including azathioprine, cyclosporine, or mycophenolate mofetil, play a pivotal role in augmenting the immunomodulatory response and minimizing steroid-related adverse effects.
- Biologic therapies, such as tumor necrosis factor-alpha (TNF-α) inhibitors or interleukin-6 (IL-6) antagonists, offer targeted immunomodulation in refractory cases or steroid-dependent individuals.
- Surgical interventions, encompassing vitrectomy or cataract extraction, may be warranted to address complications like vitreous opacities or cataract formation.
Prognosis and Complications
The prognosis of VKH syndrome hinges on early recognition, prompt initiation of therapy, and diligent monitoring to forestall disease progression and mitigate complications. While many patients achieve remission and visual recovery with aggressive treatment, delays in diagnosis or inadequate therapy may herald sight-threatening complications, including irreversible vision loss, secondary glaucoma, retinal detachment, or cataract formation. Long-term vigilance and interdisciplinary collaboration are imperative to optimize outcomes and minimize the burden of VKH syndrome on affected individuals.
Conclusion
Vogt-Koyanagi-Harada syndrome epitomizes the intricate interplay between immune dysregulation and multi-organ involvement, posing diagnostic dilemmas and therapeutic challenges. By elucidating its clinical nuances, deciphering its pathogenesis, and delineating optimal treatment strategies, healthcare providers can navigate the complexities of VKH syndrome with precision and compassion. Ongoing research endeavors hold promise for unraveling the enigmatic facets of this rare autoimmune disorder, paving the way for enhanced diagnostic modalities, targeted therapies, and improved clinical outcomes in the quest to conquer Vogt-Koyanagi-Harada syndrome.
World Eye Care Foundation’s eyecare.live brings you the latest information from various industry sources and experts in eye health and vision care. Please consult with your eye care provider for more general information and specific eye conditions. We do not provide any medical advice, suggestions or recommendations in any health conditions.
Commonly Asked Questions
Yes, VKH syndrome may recur in some individuals, particularly if treatment is discontinued prematurely or if there is inadequate control of the autoimmune response. Long-term surveillance and regular follow-up care are essential to monitor for disease recurrence and adjust treatment as needed.
While there are no specific preventive measures for VKH syndrome, maintaining overall ocular health and minimizing exposure to potential triggers, such as viral infections or environmental stressors, may help reduce the risk of disease onset or exacerbation.
Yes, VKH syndrome has the potential to cause irreversible vision loss and other sight-threatening complications if left untreated or inadequately managed. Prompt diagnosis and initiation of appropriate therapy are crucial for preserving vision and preventing long-term sequelae.
While VKH syndrome has a global distribution, it appears to be more prevalent in regions with higher UV radiation exposure, such as tropical and subtropical areas. However, cases have been reported in diverse climates worldwide.
Yes, diagnostic criteria for VKH syndrome typically encompass a combination of clinical findings, ophthalmic examination, and ancillary tests. These may include the Revised Diagnostic Criteria for Vogt-Koyanagi-Harada Disease, formulated by the International Committee on Vogt-Koyanagi-Harada Disease.
Skin involvement in VKH syndrome often manifests as depigmentation or hypopigmented patches, particularly affecting sun-exposed areas like the face and extremities. This phenomenon, known as vitiligo-like depigmentation, is a hallmark feature of the disease.
Vogt-Koyanagi-Harada syndrome can affect individuals of all ages, including children. While it predominantly manifests in adults aged 20 to 50 years, pediatric cases have been reported, albeit less frequently.
While the exact triggers remain elusive, viral infections, particularly Epstein-Barr virus (EBV), have been implicated as potential precipitating factors for VKH syndrome. Trauma, stress, and other environmental factors may also play a role in triggering the autoimmune response.
Yes, certain human leukocyte antigen (HLA) alleles have been implicated in predisposing individuals to Vogt-Koyanagi-Harada syndrome. Specific HLA haplotypes, such as HLA-DR4 and HLA-DRB1*0405, are known to confer increased susceptibility to the disease.
Vogt-Koyanagi-Harada syndrome is considered rare, with varying prevalence rates reported across different populations. It appears to occur more frequently in certain ethnic groups, particularly individuals of Asian, Native American, Hispanic, or Middle Eastern descent.



news via inbox
Subscribe here to get latest updates !